
酶
酶·
酶通论·
酶的概念·
酶是具有催化活性的蛋白质/核酸,也叫生物催化剂。
酶促反应是酶催化的反应;其中发生化学变化的物质叫底物。
酶催化的特点
- 不参与反应,不改变反应的平衡常数
- 酶降低活化能,加速反应速率
- 酶易失活
- 催化效率很高
- 高度专一性
- 酶的活力可控
- 酶浓度(诱导/抑制酶合成;调节酶降解)
- 激素调节酶活性
- 反馈抑制调节酶活性
- 抑制剂和激活剂调节
- 共价修饰调节
- 别构调节
- 酶原激活
- 同功酶
酶的化学组成
- 单纯蛋白质酶类
- 结合蛋白质酶类
- 酶蛋白
- 辅助因子(金属离子、金属有机物、小分子有机物)
- 辅酶(辅酶I,II)非共价键结合
- 辅基(FAD、FMN)共价键结合
酶的分子特点
- 单体酶(溶菌酶)
- 寡居酶(乳酸脱氢酶)
- 多酶复合体(丙酮酸脱氢酶系)
酶的分类·
- 习惯名称
- 根据底物
- 根据催化反应性质
- 系统名称
标明底物(两个以上用冒号隔开)及催化反应性质等
- 酶的六大类
- 氧化还原类
包括脱氢酶、氧化酶、过氧化物酶等
$\ce{2A·2H + O_2 <=>[酶] 2A + 2H_2O}$
$\ce{A·2H + O_2 <=>[酶] A + H_2O_2}$
例:邻二苯酚的氧化反应
$\ce{A·2H + B <=>[酶] A + B·2H}$
例:乳酸脱氢酶 - 转移酶类
$\ce{A-R + B <=>[酶] A + B-R}$
例:谷丙转氨酶 - 水解酶类
$\ce{AB + H_2O <=>[酶] AOH + BH}$
- 裂合酶类
$\ce{AB <=>[酶] A + B}$
- 异构酶类
$\ce{A <=>[酶] B}$
- 连接酶类
- $\ce{A + B + ATP ->[酶] AB + ADP + Pi}$
- $\ce{A + B + ATP ->[酶] AB + AMP + PPi}$
- $\ce{A + B + ATP ->[酶] AB + ADP + Pi}$
- 氧化还原类
酶的专一性·
- 结构专一性
底物对酶的化学结构有要求,分为绝对专一性和相对专一性。
- 立体专一性
在结构专一性的基础上对立体异构有要求,可分旋光异构专一性和几何异构专一性。
- 为什么专一
- 锁钥假说 1894年 Fischer 不完全正确
- 诱导锲合假说 1958年 Koshland
酶与底物结合后,酶的构象发生变化,使得酶与底物更好的结合,从而促进反应的进行。
酶的活力测定与分离提纯·
- 酶活力
在一定条件下所催化的某一反应的速率(即单位时间/体积中底物减少或增加的量)
- 酶活力的测定
测定完成一定反应的时间
测定单位时间的反应量
办法- 分光光度法
要求产物底物对目标光吸收率不同
优点:简单、快速、准确、方便监测。 - 荧光法
要求产物底物具有荧光性质
优点:灵敏度高。
缺点:易被干扰。 - 同位素标记法
灵敏度最高。
- 电化学法
pH计、氧电极法等。
- 分光光度法
- 酶活力的测定
测定完成一定反应的时间
- 酶单位
一定条件下在一定时间内将一定量底物转化为产物的酶的量
单位:IU Kat
1IU = 1μmol/min (μ = 10-6)
1Kat = 1mol/s - 比活力
每单位质量酶所具有的活力单位数
单位:IU/mg 等
- 提纯
总活力损失,比活力提高。
特殊酶·
参见p4
- 核酶
rna酶
- 抗体酶
在抗体可变区有酶属性的具有催化活性的免疫球蛋白
酶促反应动力学·
研究酶促反应速率及影响其的因素
酶与底物的中间络合物学说
- 现象
- 一级反应特征低浓度的时候成正比
- 混合反应特征中浓度的时候成不正比部分比例增加
- 零级反应特征高浓度的时候达Vmax
- 猜想
- 酶首先和底物结合形成ES,然后再解离成P和S
- $\ce{ S + E <=> ES -> P + S}$
- 验证
- 电镜直接观察到了ES
米氏方程·
$V = \frac{V_{max} \cdot [S]}{K_m + [S]}$
Meichele-Menten 提出:
$\ce{S + E <=>[K_1][K_2] SE ->[K_3] P + E }$ (其中K1 >> K2 / K3)
推导出:
$\ce{V = V_{max} \frac{[S]}{K_s + [S]}}$
Briggs-Haldane 提出稳态理论:
$\ce{S + E <=>[K_1][K_2] SE <=>[K_3][K_4] P + E }$
推倒:
ES浓度变化速度:$\frac{d[ES]}{dt}$
- 在反应初期,
- K4 ~ 0,
此时 $\frac{d[ES]}{dt}$ 为 $ K_1 ( [E] - [ES] ) \cdot [S]$ - 此时 $-\frac{d[ES]}{dt}$ 为 $ K_2 \cdot [ES] + K_3 \cdot[ES] \textcircled{2}$
- K4 ~ 0,
- 在稳态下:
- $ K_1 ( [E] - [ES] ) \cdot [S] = K_2 \cdot [ES] + K_3 \cdot[ES] $
- 即:
- $\frac{k_2 + k_3}{k_1} = \frac{( [E] - [ES] ) \cdot [S]}{[ES]} \textcircled{3} , $
- 令:$K_m = \frac{k_2 + k_3}{k_1}$
- 可得:$[ES] = \frac{[E] \cdot [S]}{K_m + [S]}\textcircled{4}$
- 又:
- V = K3 [ES],
- 可得:$V = K_3 \cdot \frac{[E] \cdot [S]}{K_m + [S]}$
- V = K3 [ES],
- 当:
- [S] >> [E] 时, 有 [E] = [ES],
- 那么 $V_{max} = K_3 \cdot [E]\textcircled{7}$
- 此时有:
- $V = \frac{V_{max} \cdot [S]}{K_m + [S]}$(米氏方程)
-
所以:
- Km >> [S] 时,V = $\frac{V_{max} \cdot [S]}{K_m}$
- Km << [S] 时,V = Vmax
- Km = [S] 时,V = $\frac{V_{max}}{2}$
- 我们称Km为米氏常数(mol/L)
- Km >> [S] 时,V = $\frac{V_{max} \cdot [S]}{K_m}$
-
$\textcircled{2}$ 可以判定酶的专一性和天然底物
-
$\textcircled{3}$ 可以计算Vmax和V间的关系
-
$\textcircled{7}$ 可以判定反应方向与途径
- 注意:
- $K_m \neq K_s$,仅在 K1 >> K3 / K2 时才可近似看作 Ks,此时 $\frac{1}{K_m}$ 近似表示酶与底物的亲和力。
- $K_{max}$ 在特定的反应中是常数
- [S]很大时,$V_{max} = K_3[E]$,在这里 $K_3$(一级转换常数)即为Kcat / TN。
- 作图:
- 双倒数 $\frac{1}{v} = \frac{K_m}{V_{max}} \cdot \frac{1}{[S]} + \frac{1}{V_{max}}$
- Eadie-Hofstee法 $V = -K_m \cdot \frac{V}{[S]} + V_{max}$
- Hanes-Woolf法 $\frac{[S]}{V} = \frac{K_m}{V_{max}} + \frac{[S]}{V_{max}}$
- 直接作图 $V_{max} = V + \frac{V}{K_m \cdot [S]}$
多底物反应的酶促反应动力学·
- 双底物反应
酶的抑制作用·
指酶的必需基团变性,酶的活性下降,但不引起酶变性。
- 抑制程度的表示
- 相对活力分数 $a = \frac{v_1}{v_0}$
- 相对活力百分数 a%
- 抑制分数 1 - a
- 抑制百分数 (1 - a)%
抑制剂
- 抑制剂的分类
通过物理方法鉴别可逆与不可逆的抑制作用
- 可逆抑制剂可以通过物理方法让酶恢复活力的抑制剂
- 竞争抑制剂
化学组成与构象同底物相似,能与酶结合,变相降低有效酶浓度
- 非竞争抑制剂
酶可以与(底物和抑制剂)【不分先后】同时结合形成三元复合物,但此复合物无法进一步反应
- 反竞争抑制剂
酶要先与底物结合,才能和抑制剂结合(多见于多底物反应)
- 竞争抑制剂
- 不可逆抑制剂反之,一般通过共价键结合
- 专一抑制剂
专一地作用于某酶的活性部位的必需基团上
Kcat型抑制剂 也是 酶的底物,在酶的催化下会使酶自己的基团失活
Ks型抑制剂 与 底物结构类似,带有活泼化学基团,可修饰酶的活性基团 - 非专一抑制剂
作用在酶的一类或几类基团上
有机磷:作用于丝氨酸的羟基
有机汞/有机砷:作用于巯基
重金属盐:高浓度失活,低浓度抑制
烷化剂:使巯基、羟基、氨基、羧基 等烷基化
- 专一抑制剂
- 可逆抑制剂可以通过物理方法让酶恢复活力的抑制剂
- 抑制剂动力学
Ki 是 抑制剂常数
- 竞争性抑制
- $V = \frac{V_{max}[S]}{K_m (1 + \frac{[I]}{K_i})+ [S]}$
双倒数式:
$\frac{1}{V} = \frac{K_m}{K_{max}}(1 + \frac{[I]}{K_i}) \cdot \frac{1}{[S]} + \frac{1}{V_{max}}$
- $V = \frac{V_{max}[S]}{K_m (1 + \frac{[I]}{K_i})+ [S]}$
- 非竞争性抑制
- $V = \frac{V_{max}[S]}{(K_m + [S])(1 + \frac{[I]}{K_i})}$
双倒数式:
$\frac{1}{V} = \frac{K_m}{K_{max}} (1 + \frac{[I]}{k_i})\frac{1}{[S]} + \frac{1}{V_{max}}(1 + \frac{[I]}{K_i})$
- $V = \frac{V_{max}[S]}{(K_m + [S])(1 + \frac{[I]}{K_i})}$
- 反竞争性抑制
- $V = \frac{V_{max}[S]}{K_m + [S] \cdot (1 + \frac{[I]}{K_i})}$
双倒数式:
$\frac{1}{V} = \frac{K_m}{K_{max}} \frac{1}{[S]} + \frac{1}{V_{max}}(1 + \frac{[I]}{K_i})$
- $V = \frac{V_{max}[S]}{K_m + [S] \cdot (1 + \frac{[I]}{K_i})}$
- 竞争性抑制
环境等因素对酶促反应的影响
- 温度 温度系数Qn:指每增加n℃,前后反应速率之比
- pH
酶有最适pH,大多数酶的最适pH在5~8之间。(动物:6.8~8.0)(植物微生物:4.8~6.5)
原因:- 过酸过碱 影响 空间构象
- 影响络合物解离
- 影响底物解离
- 影响有关基团解离
- 解离其他基团造成空间构象改变
- 激活剂
可以提高反应速率的物质
- 无机离子
- 小分子有机物
- 某些酶(激活酶原的激酶)
酶的作用机理及调节·
- 酶的活性部位
- 功能区域
- 结合部位专一性
- 催化部位催化反应性质
- 特点
- 占比小
- 三维实体
- 不完全锲合(诱导)
- 位于酶分子表面一个裂隙内
- 结合底物靠较弱的次级键
- 有一定的柔性与运动性
- 功能区域
- 酶的基团
- 必需基团经过化学修饰后会使整个酶失活的基团
- 非必需基团修饰后不影响酶活性
- 研究活性部位的方法
-
酶侧链基团的化学修饰法
鉴别修饰剂是否与酶的活性部位基团结合:
- 酶活力丧失速率与修饰剂浓度正相关
- 底物/与活性部位结合的可逆抑制剂可保护酶免受修饰剂的抑制
修饰形式:
- 非特异性共价修饰
- 特异性共价修饰如DFP作用于胰凝乳蛋白酶Ser195
- 亲核标记
-
动力学参数测定法
-
X射线晶体衍射法
-
定点诱变法
一个个残基改变后测试
-
酶的催化机理·
-
影响酶催化效率的因素
- 邻近效应
酶与底物间存在的亲和性会使之靠近,因此酶活性中心附近的底物浓度会增加
- 定向效应
底物向酶的活性中心靠近时,会诱导酶的构象向 底物反应基团 与 酶活性中心 正确排列的方向转变
酶的某些基团/离子 会使 底物敏感键中的基团的电子云密度 向 更敏感的状态 去 升高/降低。此时底物更接近过渡态。
- 邻近效应
-
酸碱催化
质子酸提供部分电子/质子碱接受部分电子 以 降低活化能。
酶中可作广义酸/碱的基团:Glu Asp Lys Arg Cys His Ser Tyr -
共价催化
亲核/亲电子催化 反应时分别 放出/获得 电子。作用于底物中心 形成中间复合物 降低活化能。(辅酶/辅基 可参加(如:磷酰酶 酰化酶 葡糖酶))
亲核基团:His Cys Asp Ser 巯基 羧基 羟基 咪唑基
亲电子基团:H+ Mg2+ Mn2+ Fe3+ -
金属离子催化
金属酶 含 紧密结合 的 金属离子
金属-激活酶 含 松散结合 的 金属离子
作用原理: 1.传递电子 (许多氧化还原酶含Cu2+ Fe3+) 2.电荷屏蔽以促进反应 (许多激酶底物为ATP-Mg2+复合物) 3.促进水的离子化 以 促进亲核催化 -
多元催化与协同效应
几个基元配合作用
如:胰凝乳蛋白酶Ser195Asp102His57构成电荷中继网 -
活性部位的微环境
底物分子与酶活性部位结合,此时会形成疏水环境,因此,微环境极性降低,底物分子 与 酶活性部位 间 静电作用力 增强。 利于酶的催化反应。
酶的催化反应实例·
-
Lysozyme溶菌酶切NAG氮乙酰氨基葡萄糖 人们发现:
[s](10-4mol/L) 相对水解率 结构 (NAG)2 0 A-B (NAG)3 1 A-B-C (NAG)4 8 A-B-C-D (NAG)5 4000 A-B-C-D-E (NAG)6 30000 A-B-C-D-E-F (NAG)8 30000 - 可得:
Lysozyme切割 D-E 间的键效率最高
在真实细胞壁中,NAG与NAM交替排列,因此:
Lysozyme作用于NAG-NAM-NAG-(NAM-NAG)-NAM
进一步:
Lysozyme 形如 凹 ,中间的疏水裂缝恰好能容纳以上寡糖。
而Lysozyme发挥作用的氨基酸是Asp52 Glu35。
Glu35上的质子转移到E环糖苷键上,使之断裂,此时,D环上的碳原子为C2+,Asp52上的O-能暂时稳定C+,其后,OH-与H+分别与C2+和Glu35侧链结合,水解完成。
-
Ser蛋白酶家族
- 胰蛋白酶 胰凝乳蛋白酶 弹性蛋白酶
- 枯草杆菌蛋白酶 凝血酶 纤溶酶
- tPA 乙酰胆碱酯酶
以上三种酶虽然一级结构不太相似,但三维结构相似。
催化三连体:His57(酸碱催化担当) Asp102(定位His57) Ser195(共价结合肽键 使其断裂)
Ser羟基转移一个质子给His咪唑基上的N,使之带正电,可以与带负电的Asp静电稳定。此时,Ser羟基的O带负电,亲核攻击羰基上的C,形成共价化合物。(酰基酶)
其后,His57N上的质子流向酰胺键上的N,形成不稳定化合物,发生肽键断裂,氨基产物释放。
再后,H20发动亲核攻击,OH 接 原氨基酸 的 C,H 接 His57 Ser195。于是,四面体瓦解,释放 羧基产物。核酸催化略(二轮复习再学)
酶活性的调节控制·
- 别构调节
-
概念多亚基酶分子的非催化部位与某些化合物非共价结合,使酶的构象发生变化,从而影响酶的活性。(别构酶/变构酶)
使 酶别构 的物质 为 别构剂/效应物,一般是 小分子代谢物/辅助因子
正效应物/别构激活剂, 负效应物/别构抑制剂
酶上有和 底物/调节物 结合的 部位,这 两个 部位可以在 同一亚基 或 不同亚基 上- 同促效应
活性部位 和 调节部位 相同
- 异促效应
活性部位 和 调节部位 不同。但,可能相互影响产生协同效应如:天冬氨酸转氨甲酰酶
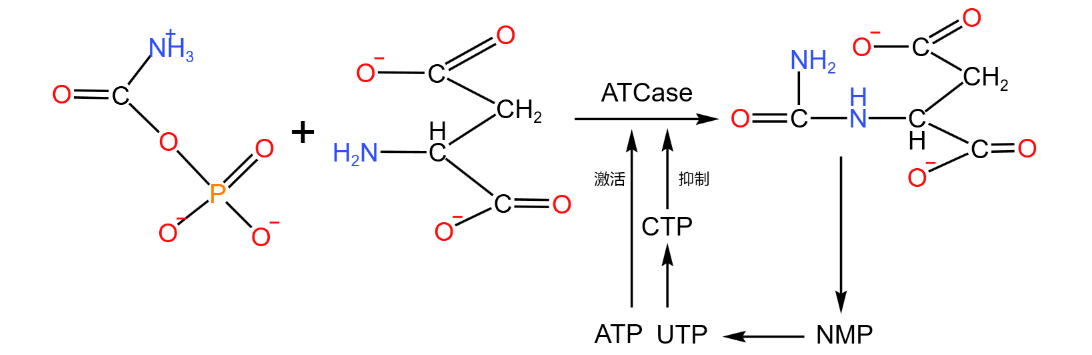
ATCase 亚基排列 3R22C3
- 同促效应
-
酶促反应动力学不符合米氏方程
[S]对V ,正协同 S形 ,负协同 表观双曲线。
对 正协同 来说 [S]对V求导,导数大
对 正协同 来说 [S]对V求导,[S]在0附近的导数大,但随即导数下降到接近0 Kashland标准协同指数(CI) 又 饱和比数(RS)
$\frac{ 结合位点饱和90%时[S] }{ 结合位点饱和10%时[S] } = 81^{ \frac{ 1 }{ n } }$
其中,n为 Hill系数。n>1:正协同效应别构酶,n<1:负协同效应别构酶,n=1:米氏酶 -
调节机制
- 齐变模型(MWC)对称亚基 -> 齐步变化 -> 对称亚基
- 别构酶是 由确定数目 亚基 组成的 寡聚酶, 众亚基平等 ,有对称轴
- 每个亚基 对 一种调节物 只有一个 结合位点
- 每种亚基都有 紧密构象 与 松弛构象
- 构象状态改变时 对称性不变
- 序变模型(KNF)全T -> 有T有R -> 全R
- 配体不存在时,酶的构象为T(紧密构象)
- 按序依次变化
- 亚基间可以是正/负协调 作用
- 齐变模型(MWC)对称亚基 -> 齐步变化 -> 对称亚基
-
- 酶原激活
$\ce{ 酶原 ->[水解酶][不可逆] 酶 }$
胰蛋白酶原的转化: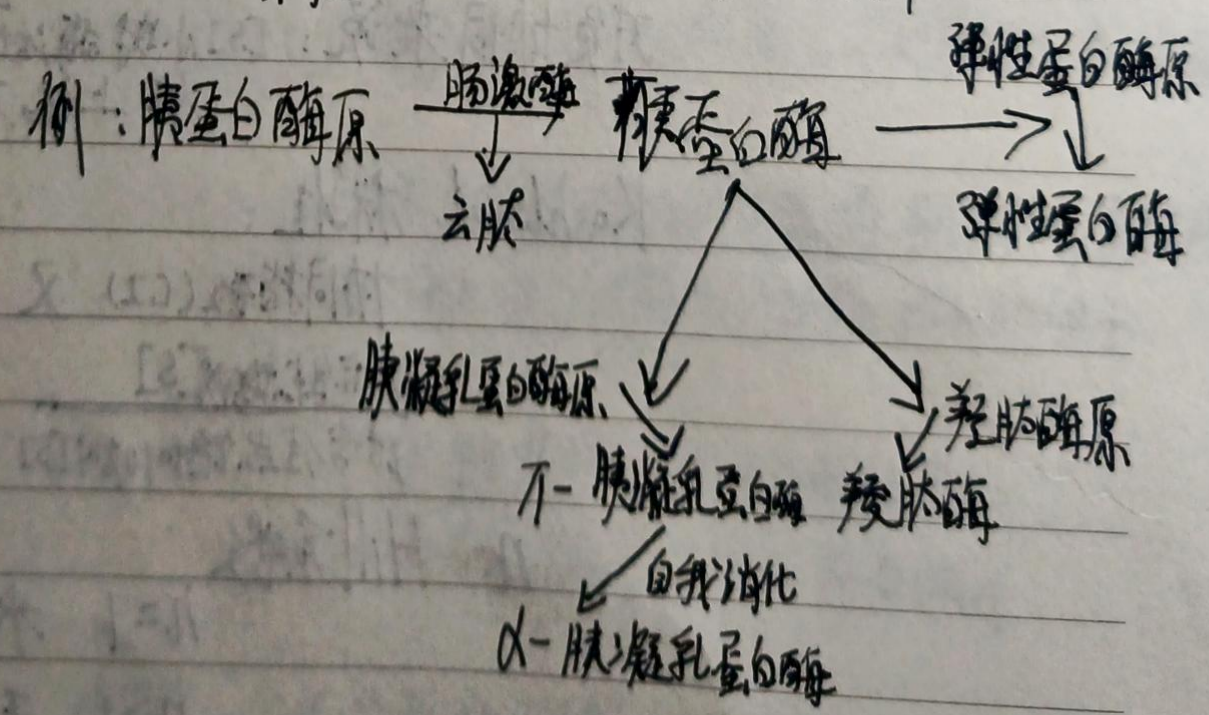
凝血机制
-
可逆共价修饰 共价修饰酶 可通过 其他酶 共价修饰 己身多肽链上的 一些基团 来 调节活性
特点:- 反应灵敏
- 节约能量
- 机制多样
- 常受激素/神经之指令
- 级联放大,高效快捷
例如:
-
磷酸化修饰(重点)
- $\ce{ 蛋白质 <=>[ ATP -> ADP 蛋白激酶 ][ nPi <- H_2O 蛋白磷酸酶 ] 蛋白质-nP }$
两类蛋白激酶:
- Ser/Thr羟基蛋白激酶
- Tyr羟基磷酸化蛋白激酶
三类蛋白磷酸酶:
- Ser/Thr蛋白磷酸酶
- Tyr蛋白磷酸酶
- 两重底物型蛋白磷酸酶
例如:
蛋白激酶A的激活(cAMP依赖)(R2C2,每个R上有两个cAMP位点)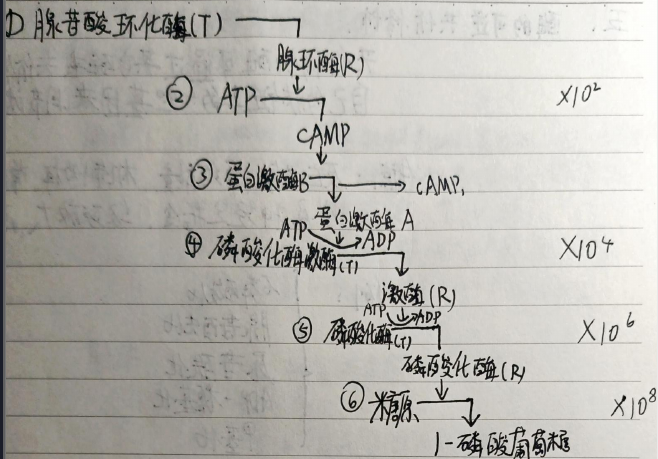
-
腺苷酰化修饰
-
尿苷酰化修饰
-
ADP-糖基化修饰
-
甲基化修饰
-
同工酶isoenzyme 具有不同分子形式 但 催化相同化学反应 的一组酶
意义:- 遗传标志
- 和个体发育 分化 密切相关
- 适应不同代谢需求而产生的差异
例如:
- 乳酸脱氢酶(LHD)由 4个 2种不同亚基 组成。因此 有 $ C^2_4 - 1 = 5 $ 种LHD,即H4 ~ M4。
不同组织的细胞中LHD1 ~ LHD5 含量不同。可诊断疾病。 - 大肠杆菌中的 天冬氨酸激酶 的 同工酶
考点总结及名词解释·
有待补充